
Integrated Assessment Services helps organizations obtain 510K premarket notifications to the US FDA. A premarket notification, also called a “premarket submission” or “510(k),” is used by medical device manufacturers to show the FDA that their device is substantially equivalent (as “” A “predictive” or “reference”) device is another. A legally marketed device is already on the market. It’s easier for your company to clean up its medical devices by providing significantly less evidence from clinical trials. Makes it faster. The 510(k) premarket notification process was established to help speed the introduction of new devices that are “substantially equivalent” to devices that are already legally authorized for the same use. have been marketed for the basic.
What is a 510(k)?
A 510(k) premarket notification is used by medical device manufacturers to legally market a new medical device. This process requires the Food and Drug Administration (FDA) to submit technical information on a predetermined form called a premarket notification [510(k)] application, which is reviewed by FDA scientists. Tested for effectiveness and labeling. Let’s review.
Some examples of devices that may be cleared through 510(k) include, but are not limited to:
- General fitness equipment
- Diagnostic imaging equipment (MRI, XRAY, etc.)
- Collecting and processing blood samples
- Surgical instruments (laparoscopy, staplers, etc.)
- Devices for treating and diagnosing illness,disease or injury.
Some devices require the FDA to comply with more stringent regulations before marketing. In these cases, the medical device manufacturer must obtain an Investigational Device Exemption (IDE) prior to clinical trials in humans or submit a Primary Device Exemption (PMA) for human’s uses devices.
Many medical device manufacturers have questions about the process for bringing a Class II or high-risk Class III device to market in the United States, including those that use international sales subsidiaries in the United States or import devices. do For example, how can a company bring its foreign-made device to American patients? How do companies get FDA approval for their device and legally market it in the US?
Validity:
There is no set term for a 510(k). If you have an existing 510(k) on file with FDA and want to include new devices in this submission, you can use your original document as long as you have all the required information on file. . Documents are available. Information is available.
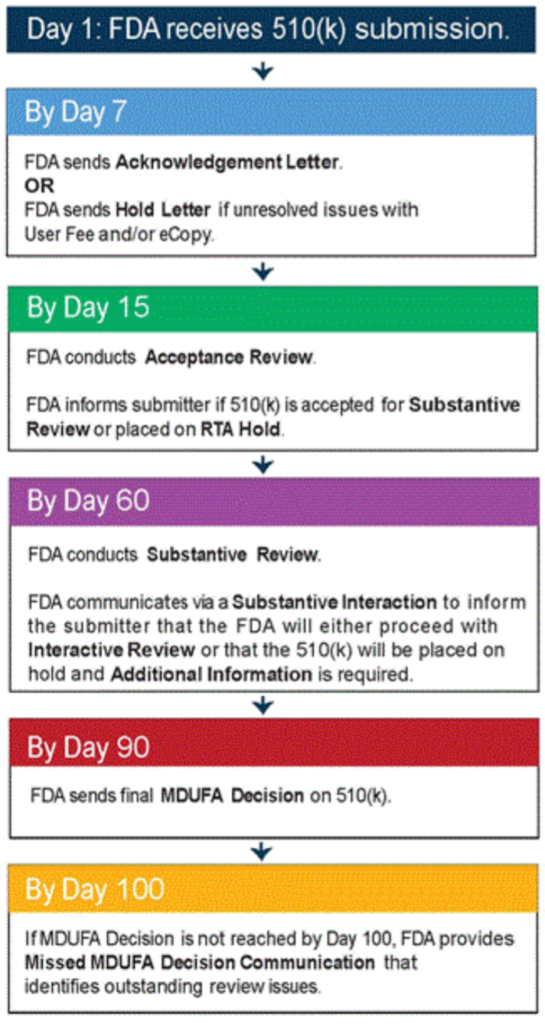
When you submit a 510(k) to the FDA, the agency’s Center for Devices and Radiological Health (CDRH) will review it within 90 days of submission to determine whether it is complete. The agency may also issue a “Not Sufficiently Equivalent” (NSE) letter if omissions or errors are found in your 510(k).